Thermal transport
Thermoelectric materials, which can both generate electricity from waste heat and use electricity for solid-state Peltier cooling, are to date inefficient, compared to conventional generators and refrigerators. One way to obtain systems with improved efficiency is to engineer nanostructured semiconductors, so as to reduce the thermal conductivity of the crystalline materials while preserving their electronic properties. Such a strategy has been recently applied to several semiconductors with promising results.
We are developing simulation frameworks to investigate thermal transport in nanostructured materials, that are based on molecular dynamics simulations and the Boltzman transport equation.
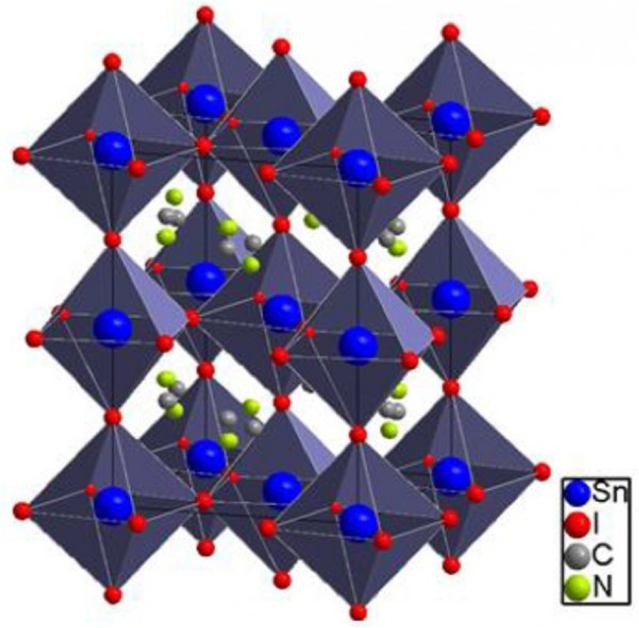 Using ab initio calculations, we showed that hybrid organic/inorganic CH3NH3AI3 (A = Pb and Sn) perovskites may be promising materials for solar thermoelectric applications. We found that their large carrier mobilities mainly originate from a combination of the small effective masses of electrons and holes and a relatively weak carrier–phonon interaction. We propose that by tuning the carrier concentration to values of the order of ∼1018cm–3, the thermoelectric figure of merit of Sn and Pb based perovskites may reach values ranging from 1 to 2.
Using ab initio calculations, we showed that hybrid organic/inorganic CH3NH3AI3 (A = Pb and Sn) perovskites may be promising materials for solar thermoelectric applications. We found that their large carrier mobilities mainly originate from a combination of the small effective masses of electrons and holes and a relatively weak carrier–phonon interaction. We propose that by tuning the carrier concentration to values of the order of ∼1018cm–3, the thermoelectric figure of merit of Sn and Pb based perovskites may reach values ranging from 1 to 2.
-
"Theoretical Study of Heat Transport in Si-based Ordered, Disordered and Nanostructured Bulk Materials",
Yuping He, Ph.D. Thesis (2011)
-
"Perovskites for Solar Thermoelectric Applications: a First Principle Study of CH3NH3AI3 (A=Pb and Sn)",
Yuping He and Giulia Galli,
Chem. Mater. 26, 5394 (2014)
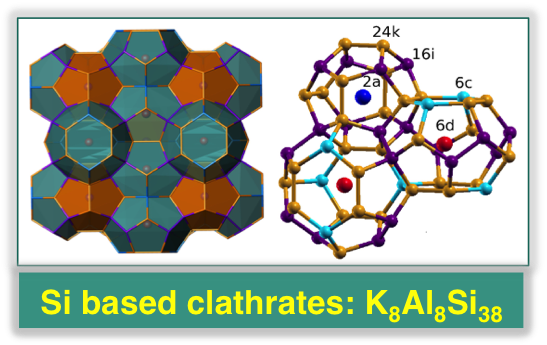 Using first-principles calculations, we investigated the thermoelectric properties of a newly synthesized Si-based ternary clathrate K8Al8Si38, composed of ∼1 nm hollow cages with a metal atom inside. We found that, similar to other nanostructured type I clathrates, this system is a semiconductor and has a low thermal conductivity (∼1 W/mK). It was long believed that the mere presence of rattling centers was responsible for the low lattice thermal conductivity of type I clathrates. We found instead that the cage structural disorder induced by atomic substitution plays a crucial role in determining the conductivity of these materials, in addition to the dynamics of the guest atoms. Our calculations showed that the latter is substantially affected by the charge transfer between the metal and the cages.
Using first-principles calculations, we investigated the thermoelectric properties of a newly synthesized Si-based ternary clathrate K8Al8Si38, composed of ∼1 nm hollow cages with a metal atom inside. We found that, similar to other nanostructured type I clathrates, this system is a semiconductor and has a low thermal conductivity (∼1 W/mK). It was long believed that the mere presence of rattling centers was responsible for the low lattice thermal conductivity of type I clathrates. We found instead that the cage structural disorder induced by atomic substitution plays a crucial role in determining the conductivity of these materials, in addition to the dynamics of the guest atoms. Our calculations showed that the latter is substantially affected by the charge transfer between the metal and the cages.
-
"Nanostructured Clathrate Phonon Glasses: Beyond the Rattling Concept",
Yuping He and Giulia Galli,
Nano Lett. 14, 2920 (2014)
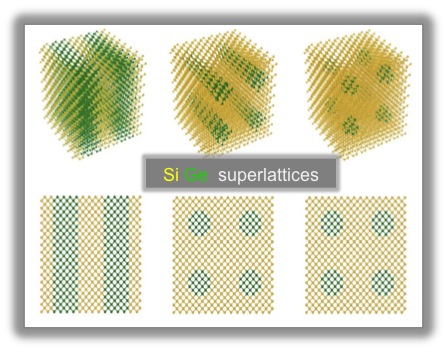 We computed the thermal conductivity of planar superlattices, arrays of Ge nanowires and nanodots embedded in Si, by using a fully atomistic Monte Carlo solution of the Boltzmann transport equation, and we investigated how dimensionality affects heat transport in Si-Ge superlattices.
We also investigated thermal transport in thin SiGe wires and in nanoporous SiGe, using molecular dynamics and lattice dynamics calculations, and we investigated how morphology affects changes in the thermal conductivity of SiGe at the nanoscale.
We computed the thermal conductivity of planar superlattices, arrays of Ge nanowires and nanodots embedded in Si, by using a fully atomistic Monte Carlo solution of the Boltzmann transport equation, and we investigated how dimensionality affects heat transport in Si-Ge superlattices.
We also investigated thermal transport in thin SiGe wires and in nanoporous SiGe, using molecular dynamics and lattice dynamics calculations, and we investigated how morphology affects changes in the thermal conductivity of SiGe at the nanoscale.
-
"Dimensionality and heat transport in Si-Ge superlattices",
I.Savic, D.Donadio, F.Gygi and G.Galli,
Appl. Phys. Lett. 102, 073113 (2013)
-
"Morphology and Temperature Dependence of The Thermal Conductivity of Nanoporous SiGe",
Y. He, D. Donadio and G. Galli,
Nano Lett. 11, 3608-3611 (2011)
-
"Cluster expansion and optimization of thermal conductivity in SiGe nanowires",
M. K. Y. Chan, J. Reed, D.Doonadio, T. K. Mueller, Y. S. Meng, G. Galli and G. Ceder,
Phys. Rev. B 81, 174303 (2010)
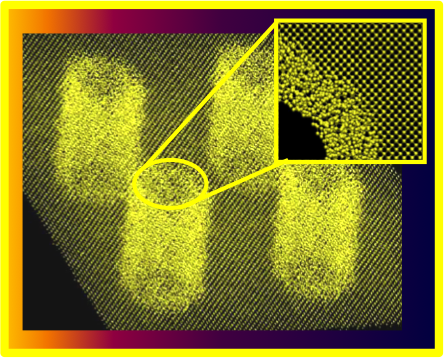 We showed that the thermal conductivity of nanoporous silicon may attain values 10−20 times smaller than in bulk Si for porosities and surface-to-volume ratios similar to those obtained in recently fabricated nanomeshes. Further reduction of almost an order of magnitude is obtained in thin films with thickness of 20 nm, in agreement with experiment. Our study was conducted using molecular dynamics simulations and lattice dynamics calculations and results were compared to those for amorphous Si, studied with the same methodologies.
We showed that the thermal conductivity of nanoporous silicon may attain values 10−20 times smaller than in bulk Si for porosities and surface-to-volume ratios similar to those obtained in recently fabricated nanomeshes. Further reduction of almost an order of magnitude is obtained in thin films with thickness of 20 nm, in agreement with experiment. Our study was conducted using molecular dynamics simulations and lattice dynamics calculations and results were compared to those for amorphous Si, studied with the same methodologies.
-
"Heat Transport in Amorphous Silicon: Interplay Between Morphology and Disorder",
Y. He, D. Donadio and G. Galli,
Appl. Phys. Lett. 98, 144101 (2011)
-
"Thermal Transport in Nanoporous Silicon: Interplay between disorder at the mesoscopic and the atomic scale",
Y. He, D. Donadio, J.-H. Lee, J. C. Grossman and G. Galli,
ACS Nano 5, 1839 (2011)
-
"Silicon stops heat in its tracks",
G. Galli and D. Donadio,
Nat. Nanotech. 5, 701 (2010)
 We carried out a series of molecular dynamics, lattice dynamics, and Boltzmann transport equation calculations aimed at understanding heat transport in Silicon nanowires. We found that the computed thermal conductivity strongly depends on the surface structure and defects present in the bulk of the wires. Our results were used to rationalize several experiments showing strong reduction of the conductivity in Si nanowires.
We carried out a series of molecular dynamics, lattice dynamics, and Boltzmann transport equation calculations aimed at understanding heat transport in Silicon nanowires. We found that the computed thermal conductivity strongly depends on the surface structure and defects present in the bulk of the wires. Our results were used to rationalize several experiments showing strong reduction of the conductivity in Si nanowires.
-
"Microscopic origin of the reduced thermal conductivity of silicon nanowires",
Yuping He and Giulia Galli,
Phys. Rev. Lett. 108, 215901 (2012)
-
"Atomistic simulations of heat transport in silicon nanowires",
D. Donadio and G. Galli,
Phys. Rev. Lett. 102, 195901 (2009)
-
"Temperature dependence of the thermal conductivity of thin silicon nanowires"
D. Donadio and G. Galli,
Nano Lett. 10, 847 (2010)
Using first-principles calculations, we investigated the thermoelectric properties of a newly synthesized Si-based ternary clathrate K8Al8Si38, composed of ∼1 nm hollow cages with a metal atom inside. We found that, similar to other nanostructured type I clathrates, this system is a semiconductor and has a low thermal conductivity (∼1 W/mK). It was long believed that the mere presence of rattling centers was responsible for the low lattice thermal conductivity of type I clathrates. We found instead that the cage structural disorder induced by atomic substitution plays a crucial role in determining the conductivity of these materials, in addition to the dynamics of the guest atoms. Our calculations showed that the latter is substantially affected by the charge transfer between the metal and the cages.
- "Nanostructured Clathrate Phonon Glasses: Beyond the Rattling Concept", Yuping He and Giulia Galli, Nano Lett. 14, 2920 (2014)
We computed the thermal conductivity of planar superlattices, arrays of Ge nanowires and nanodots embedded in Si, by using a fully atomistic Monte Carlo solution of the Boltzmann transport equation, and we investigated how dimensionality affects heat transport in Si-Ge superlattices.
We also investigated thermal transport in thin SiGe wires and in nanoporous SiGe, using molecular dynamics and lattice dynamics calculations, and we investigated how morphology affects changes in the thermal conductivity of SiGe at the nanoscale.
- "Dimensionality and heat transport in Si-Ge superlattices", I.Savic, D.Donadio, F.Gygi and G.Galli, Appl. Phys. Lett. 102, 073113 (2013)
- "Morphology and Temperature Dependence of The Thermal Conductivity of Nanoporous SiGe", Y. He, D. Donadio and G. Galli, Nano Lett. 11, 3608-3611 (2011)
- "Cluster expansion and optimization of thermal conductivity in SiGe nanowires", M. K. Y. Chan, J. Reed, D.Doonadio, T. K. Mueller, Y. S. Meng, G. Galli and G. Ceder, Phys. Rev. B 81, 174303 (2010)
We showed that the thermal conductivity of nanoporous silicon may attain values 10−20 times smaller than in bulk Si for porosities and surface-to-volume ratios similar to those obtained in recently fabricated nanomeshes. Further reduction of almost an order of magnitude is obtained in thin films with thickness of 20 nm, in agreement with experiment. Our study was conducted using molecular dynamics simulations and lattice dynamics calculations and results were compared to those for amorphous Si, studied with the same methodologies.
- "Heat Transport in Amorphous Silicon: Interplay Between Morphology and Disorder", Y. He, D. Donadio and G. Galli, Appl. Phys. Lett. 98, 144101 (2011)
- "Thermal Transport in Nanoporous Silicon: Interplay between disorder at the mesoscopic and the atomic scale", Y. He, D. Donadio, J.-H. Lee, J. C. Grossman and G. Galli, ACS Nano 5, 1839 (2011)
- "Silicon stops heat in its tracks", G. Galli and D. Donadio, Nat. Nanotech. 5, 701 (2010)
We carried out a series of molecular dynamics, lattice dynamics, and Boltzmann transport equation calculations aimed at understanding heat transport in Silicon nanowires. We found that the computed thermal conductivity strongly depends on the surface structure and defects present in the bulk of the wires. Our results were used to rationalize several experiments showing strong reduction of the conductivity in Si nanowires.
- "Microscopic origin of the reduced thermal conductivity of silicon nanowires", Yuping He and Giulia Galli, Phys. Rev. Lett. 108, 215901 (2012)
- "Atomistic simulations of heat transport in silicon nanowires", D. Donadio and G. Galli, Phys. Rev. Lett. 102, 195901 (2009)
- "Temperature dependence of the thermal conductivity of thin silicon nanowires" D. Donadio and G. Galli, Nano Lett. 10, 847 (2010)