We are developing ab initio methods based on density functional and many body perturbation theory to compute optoelectronic properties of materials and to predict electronic spectra. We have released the WEST code, a massively parallel software for large scale electronic structure calculations based on many-body perturbation theory.
"Implementation and Validation of Fully-Relativistic GW Calculations: Spin-Orbit Coupling in Molecules, Nanocrystals and Solids",
P. Scherpelz, M. Govoni, I. Hamada, and G. Galli ,
J. Chem. Theory Comput.12 (8), 3523-3544 (2016)
The WEST Data Collections provide open web-based access to electronic structure properties computed with WEST. Explore the GW100 set.
GW and BSE calculations
We have developed ab initio approaches to compute optical absorption and
emission spectra of molecules and solids, which are suitable for the study of
large systems and give access to spectra within a wide energy range. For
absorption spectra, the quantum Liouville equation is solved iteratively within
first order perturbation theory, with a Hamiltonian containing a static self-energy
operator. For emission spectra, we use a spectral decomposition of the static
dielectric matrix as a basis for the frequency dependent density-density response
function. Explicit calculations of single particle excited states and inversion and
storage of dielectric matrices
are avoided using techniques based on Density Functional Perturbation Theory.
"Implementation and Validation of Fully-Relativistic GW Calculations: Spin-Orbit Coupling in Molecules, Nanocrystals and Solids",
P. Scherpelz, M. Govoni, I. Hamada, and G. Galli ,
J. Chem. Theory Comput. (2016), accepted
"GW calculations using the spectral decomposition of the dielectric matrix: Verification, validation, and comparison of methods",
T.A. Pham, H.-V. Nguyen, D. Rocca and G. Galli,
Phys. Rev. B87, 155148 (2013)
"Predictive Theory and Modelling of Heterogeneous Interfaces", Tuan Anh Pham, Ph.D. Thesis (2014)
"Spectral representation analysis of dielectric screening in solids and molecules",
A. Kaur, E.R. Ylvisaker, D. Lu, T.A. Pham, G. Galli and W.E. Pickett,
Phys. Rev. B87, 155144 (2013)
"Electronic excitations in light absorbers for photoelectrochemical energy conversion: first principles calculations based on many body perturbation theory",
Y. Ping, D. Rocca and G. Galli,
Chem. Soc. Rev.42, 2437 (2013)
"A block variational procedure for the iterative diagonalization of non-Hermitian random-phase approximation matrices",
D. Rocca, Z. Bai, R. Li and G. Galli,
J. Chem. Phys.136, 034111 (2012)
"Ab initio calculations of optical absorption spectra: Solution of the Bethe-Salpeter equation within density matrix perturbation theory",
D. Rocca, D. Lu, and G. Galli,
J. Chem. Phys.133, 164109 (2010)
"Solution of the Bethe-Salpeter equation without empty electronic states: application to the absorption spectra of bulk systems",
D. Rocca, Y. Ping, R. Gebauer and G. Galli,
Phys. Rev. B.85, 045116 (2012)
"Improving accuracy and efficiency of calculations of photoemission spectra within many body perturbation theory",
H. Nguyen, T. Pham, D. Rocca and G. Galli,
Phys. Rev. B.85, 081101(R) (2012)
 The WEST Data Collections provide open web-based access to electronic structure properties computed with WEST. Explore the GW100 set.
The WEST Data Collections provide open web-based access to electronic structure properties computed with WEST. Explore the GW100 set.
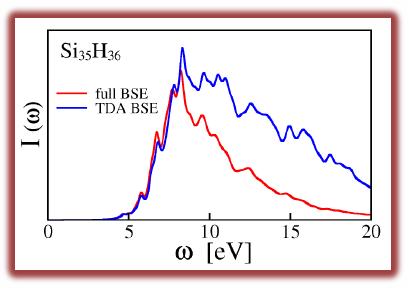 We have developed ab initio approaches to compute optical absorption and
emission spectra of molecules and solids, which are suitable for the study of
large systems and give access to spectra within a wide energy range. For
absorption spectra, the quantum Liouville equation is solved iteratively within
first order perturbation theory, with a Hamiltonian containing a static self-energy
operator. For emission spectra, we use a spectral decomposition of the static
dielectric matrix as a basis for the frequency dependent density-density response
function. Explicit calculations of single particle excited states and inversion and
storage of dielectric matrices
are avoided using techniques based on Density Functional Perturbation Theory.
We have developed ab initio approaches to compute optical absorption and
emission spectra of molecules and solids, which are suitable for the study of
large systems and give access to spectra within a wide energy range. For
absorption spectra, the quantum Liouville equation is solved iteratively within
first order perturbation theory, with a Hamiltonian containing a static self-energy
operator. For emission spectra, we use a spectral decomposition of the static
dielectric matrix as a basis for the frequency dependent density-density response
function. Explicit calculations of single particle excited states and inversion and
storage of dielectric matrices
are avoided using techniques based on Density Functional Perturbation Theory.