Vibrational spectroscopy is an ideal tool to probe the complex structure of hydrogen bonded systems, in particular ice, water and aqueous solutions. However, the interpretation of experimental spectra is usually not straightforward, due to complex spectral features associated with different bonding configurations present in these systems. Therefore, accurate theoretical predictions are required to assign spectral signatures to specific structural properties and hence to fully exploit the potential of vibrational spectroscopies.
We develop and use first-principles electronic structure methods for the simulation of vibrational
spectra of aqueous systems, including IR, Raman and sum frequency generation spectra.
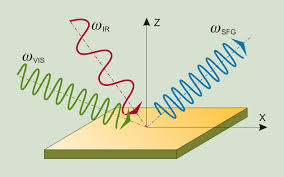
Sum Frequency Generation Spectra
We developed a first-principles framework to compute sum-frequency generation (SFG) vibrational spectra of semiconductors and insulators. The method is based on Density Functional Theory and the use of Maximally Localized Wannier functions to express the response to electric fields, and it includes the effect of electric field gradients at surfaces. In addition, it includes quadrupolar contributions to SFG spectra, thus enabling the verification of the dipolar approximation, whose validity determines the surface specificity of SFG spectroscopy.
"First-principles framework to compute sum-frequency generation vibrational spectra of semiconductors and insulators",
Quan Wan and Giulia Galli,
Phys. Rev. Lett.115, 246404 (2015)
"First principles simulations of vibrational spectra of aqueous systems", Quan Wan, Ph.D. Thesis (2015)
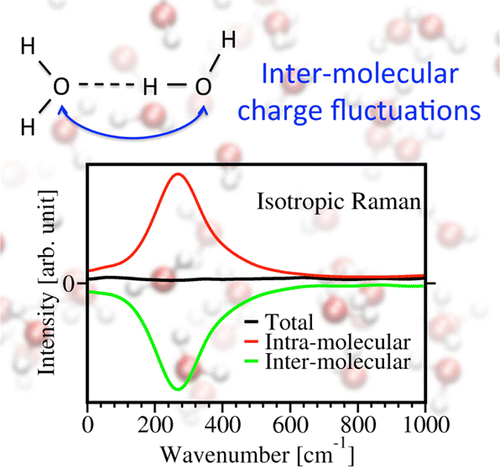
Raman Spectra
By combining first principles molecular dynamics and density functional perturbation theory, we computed the Raman spectra of liquid water from first principles. Our computed spectra are in good agreement with experiments, especially in the low frequency region. We also devised a systematic strategy to analyze the Raman intensities, which is of general applicability to molecular solids and liquids, and it is based on maximally localized Wannier functions and effective molecular polarizabilities.
"Raman Spectra of Liquid Water from Ab Initio Molecular Dynamics: Vibrational Signatures of Charge Fluctuations in the Hydrogen Bond Network",
Quan Wan, Leonardo Spanu, Giulia Galli, and François Gygi,
J. Chem. Theory Comput.9, 4124 (2013)
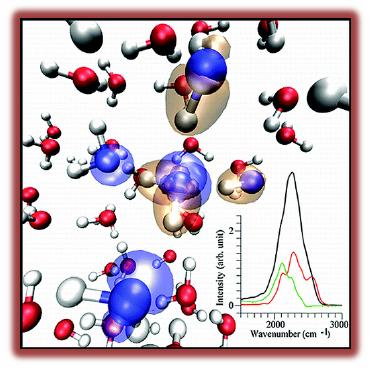
IR spectra
Ab initio MD simulations show that inter-molecular contributions play a key role in shaping the infrared (IR) spectrum of liquid water and in particular its stretching band. Hydrogen bonded (HB) and NHB molecules contribute to the IR intensities of the whole band. Ab initio MD results support the picture of a continuous dynamics in neat water, with hydrogen bonds being transient states with lifetimes of several hundred femtoseconds.
"First Principles Study of the Infrared Spectra of the Ice Ih (0001) Surface",
Tuan Anh Pham, Patrick Huang, Eric Schwegler and Giulia Galli,
J. Phys. Chem. A116, 9255 (2012)
"First Principle Analysis of the IR Stretching Band of Liquid Water",
C. Zhang, D. Donadio and G. Galli,
J. Phys. Chem. Lett.1, 1398 (2010)
"Role of dipolar correlations in the infrared spectra of water and ice",
W. Chen, M. Sharma, R. Resta, G. Galli and R. Car,
Phys. Rev. B77, 245114 (2008)